Синдром Бругада — наследственное заболевание[1], обусловленное мутацией гена SCN5A, расположенного в плече p 3-й хромосомы, кодирующего биосинтез белковых субъединиц натриевого канала кардиомиоцитов.
Впервые это понятие, позже ставшее эпонимом, предложили испано-бельгийские кардиологи — братья Педро и Хосеп Бругада[2].
Мутации генов[править | править код]
На сегодняшний день известны, по крайней мере, 5 генов, ответственных за развитие этого состояния. В зависимости от мутации гена выделяют следующие варианты:
Патогенез[править | править код]
Заболевание имеет аутосомно-доминантный тип наследования в 25 % семей. Клинические проявления синдрома Бругада развиваются обычно в молодом возрасте (до 35-40 лет), реже — наблюдаются даже в пожилом и старческом возрасте. При исследовании статистических данных, накопленных в странах Юго-Восточной Азии и Дальнего Востока, было отмечено, что в данном регионе значительно распространены случаи внезапной ночной смерти в молодом возрасте (в год от 4 до 10 случаев на 10 000 жителей, в том числе в Лаосе — 1 случай на 10 000 жителей; в Таиланде — 26-38 на 100 000).
[7]
Описаны также случаи приобретенного синдрома Бругада[2].
Синдром Бругада характеризуется наличием преходящей полной или неполной блокады правой ножки пучка Гиса, косонисходящим подъёмом сегмента S-T в правых грудных отведениях (V1-V3), рецидивирующей пароксизмальной полиморфной желудочковой тахикардией и высоким риском внезапной сердечной смерти.
Симптомокомплекс[править | править код]
Полная форма синдрома Бругада включает следующие клинико-электрокардиографические проявления:
- Типичная электрокардиографическая картина (косонисходящее повышение сегмента S-T над изолинией на 1 мм и больше в отведениях V1—V3, на некоторых ЭКГ напоминает морду бультерьера, поэтому данное изменение иногда называют «типом бультерьера»[2], спонтанное или индуцированное введением антиаритмических препаратов I класса (блокаторов натриевых каналов, например, аймалина (гилуритмала) в дозе 1 мг/кг или новокаинамида в дозе 10 мг/кг, флекаинида 2 мг/кг); полная или неполная блокада правой ножки пучка Гиса); возможно укорочение интервала Q-T и удлинение P-Q (P-R);
- Пароксизмы полиморфной желудочковой тахикардии, часто рецидивирующие;
- синкопальные (обморочные) состояния;
- высокий риск внезапной сердечной смерти вследствие полиморфной желудочковой тахикардии или фибрилляции желудочков.
Примечания[править | править код]
- ↑ Окороков А. Н., Диагностика болезней внутренних органов: Т. 10. Диагностика болезней сердца и сосудов.: — М.: Мед. лит., 2005. — 384 с.: ил. ISBN 5-89677-091-X; — ст. 239—241.
- ↑ 1 2 3 Л. А. Бокерия, О. Л. Бокерия, Л. Н. Киртбая. СИНДРОМ БРУГАДА: КЛЕТОЧНЫЕ МЕХАНИЗМЫ И ПОДХОДЫ К ЛЕЧЕНИЮ. Анналы аритмологии, № 3, 2010.
- ↑ 1 2 Antzelevitch C; Pollevick GD; Cordeiro JM; Casis, O.; Sanguinetti, M. C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A. Loss-of-Function Mutations in the Cardiac Calcium Channel Underlie a New Clinical Entity Characterized by ST-Segment Elevation, Short QT Intervals, and Sudden Cardiac Death (англ.) // Circulation (англ.)русск. : journal. — Lippincott Williams & Wilkins (англ.)русск., 2007. — Vol. 115, no. 4. — P. 442—229. — doi:10.1161/CIRCULATIONAHA.106.668392. — PMID 17224476.
- ↑ Delpon E; Cordeiro JM; Núñez L; Thomsen, P. E. B.; Guerchicoff, A.; Pollevick, G. D.; Wu, Y.; Kanters, J. K.; Larsen, C. T. Functional Effects of KCNE3 Mutation and its Role in the Development of Brugada Syndrome (англ.) // Circulation Arrhythmia and Electrophysiology : journal. — 2008. — Vol. 1, no. 3. — P. 209—218. — doi:10.1161/CIRCEP.107.748103. — PMID 19122847.
- ↑ Watanabe H; Koopmann TT; Le Scouarnec S; Yang, Tao; Ingram, Christiana R.; Schott, Jean-Jacques; Demolombe, Sophie; Probst, Vincent; Anselme, Frédéric. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans (англ.) // Journal of Clinical Investigation (англ.)русск. : journal. — 2008. — June (vol. 118, no. 6). — P. 2260—2268. — doi:10.1172/JCI33891. — PMID 18464934.
- ↑ 1 2 Bezzina, Connie R; Barc, Julien; Mizusawa, Yuka; Remme, Carol Ann; Gourraud, Jean-Baptiste; Simonet, Floriane; Verkerk, Arie O; Schwartz, Peter J; Crotti, Lia; Dagradi, Federica; Guicheney, Pascale; Fressart, Véronique; Leenhardt, Antoine; Antzelevitch, Charles; Bartkowiak, Susan; Schulze-Bahr, Eric; Zumhagen, Sven; Behr, Elijah R; Bastiaenen, Rachel; Tfelt-Hansen, Jacob; Olesen, Morten Salling; Kääb, Stefan; Beckmann, Britt M; Weeke, Peter; Watanabe, Hiroshi; Endo, Naoto; Minamino, Tohru; Horie, Minoru; Ohno, Seiko; Hasegawa, Kanae; Makita, Naomasa; Nogami, Akihiko; Shimizu, Wataru; Aiba, Takeshi; Froguel, Philippe; Balkau, Beverley; Lantieri, Olivier; Torchio, Margherita; Wiese, Cornelia; Weber, David; Wolswinkel, Rianne; Coronel, Ruben; Boukens, Bas J; Bézieau, Stéphane; Charpentier, Eric; Chatel, Stéphanie; Despres, Aurore; Gros, Françoise; Kyndt, Florence; Lecointe, Simon; Lindenbaum, Pierre; Portero, Vincent; Violleau, Jade; Gessler, Manfred; Tan, Hanno L; Roden, Dan M; Christoffels, Vincent M; Marec, Hervé Le; Wilde, Arthur A; Probst, Vincent; Schott, Jean-Jacques; Dina, Christian; Redon, Richard. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death (англ.) // Nature Genetics : journal. — 2013. — ISSN 1061-4036. — doi:10.1038/ng.2712.
- ↑ Л. М. Макаров. Синдром Бругада. журнал «Лечащий врач».
Синдром Бругада — наследственное заболевание, обусловленное мутацией гена SCN5A, расположенного в плече p 3-й хромосомы, кодирующего биосинтез белковых субъединиц натриевого каналакардиомиоцитов.
Впервые это понятие, позже ставшее эпонимом, предложили испано-бельгийские кардиологи — братья Педро и Хосеп Бругада.
Причины синдрома
Причина появления синдрома – мутация (изменение) генов, отвечающих за нормальную проницаемость клеточной оболочки кардиомиоцитов для ионизированных веществ (натрия, калия). Такие патологические мутации генов на данный момент обнаружены в нескольких хромосомах (3, 10, 11, 12, 19). Изменения, которые ими вызваны, отличаются небольшой разницей биохимических реакций.
- Читайте также:
Результатом мутации становится синдром Бругада – блокада (нарушение проводимости, сократимости, возбудимости) некоторых участков сердца.

Симптомы
Признаки синдрома Бругада на ЭКГ можно заметить с 5 лет. Манифестация симптомов происходит в возрасте 30-40 лет.
В зависимости от уровня изменений на ЭКГ при синдроме Бругада выделяют несколько клинико-электрографических типов. Полная форма включает следующие проявления:
- повышение сегмента ST над изолинией на 1 мм и выше в правых грудных отведениях, которое по форме напоминает очертания морды бультерьера (этот признак называют «типом бультерьера»);
- блокаду (полную или частичную) правой ножки пучка Гиса;
- периодическое увеличение интервала PR.
Читайте также: Вакцинация корь код мкб
Основной симптом синдрома Бругада – приступы (пароксизмы) желудочковой тахикардии, которые обычно возникают вечером и ночью. Им могут предшествовать употребление алкоголя, нагрузка или лихорадка, связанная с инфекционным заболеванием. Иногда пароксизм начинается в состоянии полного покоя. Он сопровождается:
- ощутимыми толчками в области сердца и учащением сердцебиения;
- оглушенностью;
- потливостью;
- головокружением;
- появлением «мушек» перед глазами.
Многие пациенты теряют сознание (возникает синкопе). В 89% случаев через 20-30 секунд состояние нормализуется. У остальных происходит остановка сердца из-за фибрилляции желудочков.

Диагностика
На данный момент основными способами диагностики синдрома Бругада являются:
- Читайте также:
- ЭКГ с лекарственными пробами и без;
- Холтер-ЭКГ;
- молекулярно-генетическое исследование.
Введение антиаритмических препаратов во время фармакологических проб у таких больных может вызывать желудочковую тахиаритмию (вплоть до фибрилляции желудочков), поэтому, согласно протоколу, лекарственные пробы антиаритмическими средствами проводятся только в специализированных кабинетах для электрокардиографии и при полной готовности персонала к оказанию экстренной помощи. Для проведения таких тестов могут применяться такие антиаритмические препараты:
- Новокаинамид 10 мг/кг;
- Гилуритмал 1 мг/кг;
- Флекаинид 2 мг/кг.
Методы лечения
Основной задачей терапии заболевания является недопущение приступов фибрилляции желудочков, которые и влекут за собой смерть больного. С этой целью назначается прием классических антиаритмических лекарств, которые оказывают положительное действие с примерной эффективностью в 60%. Лучшие результаты показали такие блокаторы натриевых каналов, как Ритмонорм, Энкаинид, Новокаиномид, Гилуритмал, Хинидит и т.д.
В некоторых случаях рекомендуется другие препараты (Лидокаин, Токаинид, Верапамил), действие которых менее выраженное, но вероятность побочных действий тоже меньше. Наиболее безопасным считается прием препаратов от аритмии, которые не блокируют натриевые каналы (Соталекс, Коргард), но детального изучения вопроса пока не проводилось.
При наличии в анамнезе приступов фибрилляции желудочков больному даются рекомендации по установке дефибриллятора-кардиовертера. Такие приборы не позволяют развиваться опасным для жизни видам аритмии, которые часто приводят к внезапной смерти. Если синдром Бругада обнаружен у ребенка, после имплантации прибора назначают, как правило, длительный прием препарата Хинидин, либо лекарств Дизопирамид, Пропранолол. В некоторых случаях даже прием лекарств не исключал подъема ST-сегмента по ЭКГ, в связи с чем исследования в этом вопросе должны быть продолжены. Но пока только кардиовертер-дефибриллятор достоверно предотвращает внезапную смерть больного с синдромом Бругада.
Прогноз синдрома Бругада
Прогноз синдрома Бругада неопределенный, так как степень выраженности симптомов заболевания очень вариабельна и находится в зависимости от ряда факторов. При наличии только электрокардиографических проявлений патологии без выраженных клинических симптомов прогноз относительно благоприятный. Если синдром Бругада сопровождается потерями сознания и приступами аритмии – без установки кардиовертера-дефибриллятора риск внезапной сердечной смерти возрастает во много раз. При применении данного прибора прогноз несколько улучшается, поскольку устройство может круглосуточно корректировать патологические изменения сердечного ритма.
Загрузка…
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Течение и стадии
- Диагностика
- Лечение
Названия
Название: Синдром Драве.

Синдром Драве
Описание
Синдром Драве. Это детская энцефалопатия наследственного характера, которая характеризуется эпилептиформными приступами, отставанием в психическом развитии и резистентностью к противоэпилептической терапии. Клинически заболевание проявляется полиморфными эпилептическими припадками, неврологическими расстройствами, атипическими абсансами и фокальными моторными пароксизмами. Диагностика синдрома Драве базируется на характеристике возникающих приступов, данных ЭЭГ и МРТ, идентификации мутации генов SCN1A или GABRG2. Лечение малоэффективно и проводится с целью уменьшения частоты приступов, профилактики эпилептического статуса.
Дополнительные факты
Синдром Драве или тяжелая миоклоническая эпилепсия младенчества – это аутосомно-доминантная энцефалопатия с дебютом в первые 12 месяцев жизни ребенка, которая проявляется фебрильными и афебрильными генерализованными приступами, фокальными миоклоническими пароксизмами, расстройствами неврологического статуса и дефицитом интеллекта. Впервые заболевание было описано французским психиатром и эпилептологом Шарлоттой Драве в 1978 году. Встречается данный синдром редко, распространенность – 1:20-40 тысяч детского населения. У мальчиков патология возникает вдвое чаще, чем у девочек. Исход синдрома Драве неблагоприятный – заболевание неизлечимо и слабо поддается медикаментозной терапии. Летальность составляет порядка 16-18%.
Читайте также: Стенокардия ишемическая болезнь сердца код по мкб 10
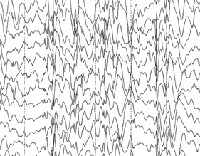
Синдром Драве
Симптомы
Клонические судороги. Миоклония. Судороги. Тремор.
Причины
Синдром Драве – это генетически детерминированная патология, которая передается по аутосомно-доминантному типу наследования. Спровоцировать развитие тяжелой миоклонической эпилепсии младенчества могут мутации локуса SCN1A на 24 участке длинного плеча 2 хромосомы (в 80% случаев) или GABRG2 на 5q34. Данные гены кодируют α1-субъединицу Na+-каналов, что приводит к нарушению физиологических процессов реполяризации и деполяризации в нейронах, и как следствие – к патологической активности ЦНС.
Течение и стадии
В клинической картине синдрома Драве выделяют 3 этапа развития: фебрильный (до 12-24 месяцев), агрессивный или катастрофический (2-8 лет), статический (старше 8 лет). Дебют заболевания происходит в возрасте от 2 месяцев до 1 года, в среднем – в 5 месяцев. До момента возникновения первых симптомов ребенок развивается нормально, неврологических и психических отклонений не наблюдается. В большинстве случаев первичными проявлениями фебрильной стадии синдрома Драве становятся фибриллярные судороги атипического характера. Они имеют большую продолжительность (свыше 20 минут), включают в себя очаговые компоненты и альтернирующие гемиконвульсии, иногда переходят в эпилептический припадок. На ранних этапах такие состояния сопровождаются субфебрильной или фебрильной температурой тела, в дальнейшем подобных проявлений не наблюдается. Зачастую при синдроме Драве приступ может быть спровоцирован гипертермией (согреванием, горячей ванной или инфекционной патологией), световыми раздражителями, резкими движениями.
Катастрофический или агрессивный период синдрома Драве характеризуется выраженными полиморфными клонико-тонико-клоническими припадками, альтернирующими гемиконвульсиями, очаговыми моторными пароксизмами, атипичными абсансами. Приступы начинаются с мышечных подергиваний по всему телу (иногда – асинхронных), переходят в кратковременную тоническую, а затем – клоническую фазы. Часто подобное состояние трансформируется в эпилептический статус, который может сохраняться до нескольких суток. В возрасте 1-2 лет у больных с синдромом Драве определяется дефицит интеллекта (олигофрения) и гиперактивность, поведенческие аномалии, нарастающие до 6-7 лет и сохраняющиеся на протяжении всей жизни. Также развиваются неврологические нарушения: мышечная гипотония, атаксия, интенционный тремор, моторная неловкость, признаки пирамидной недостаточности. В этом же возрасте у части детей возникает паттерн-сенситивность, при которой определенная одежда, обои или телевизионные передачи могут стать причиной очередного приступа.
Статическая стадия синдрома Драве характеризуется уменьшением интенсивности и частоты эпилептических припадков. Психические и неврологические отклонения остаются. Большая часть приступов возникает в ночное время или сразу после пробуждения. Как и в других периодах, они могут быть спровоцированы повышением температуры тела, ярким светом, резким движением и тд На фоне отставания в интеллектуальном развитии, нарушений психики и резистентности заболевания к лечению пациент почти полностью лишен способности адаптироваться в социуме.
 Читайте также:
Читайте также:

Диагностика
Диагностика синдрома Драве основывается на анамнестических данных, физикальном обследовании, лабораторных и инструментальных методах исследования. Из анамнеза педиатром выясняется возраст, в котором произошла манифестация патологии, первичные проявления, характеристика приступов, степень их тяжести и динамика развития. При осмотре ребенка в межприступный период можно выявить отставание в интеллектуальном развитии (ЗПР), гиперактивность, нарушения неврологического статуса. Во время припадка определяются атипичные абсансы, очаговые расстройства, альтернирующие гемиконвульсии.
Общие лабораторные анализы (ОАК, ОАМ, анализ кала) малоинформативны – выраженные отклонения от возрастной нормы, как правило, отсутствуют. Из инструментальных методов исследования при синдроме Драве используются электроэнцефалограмма (ЭЭГ) и магнитно-резонансная томография (МРТ). Между приступами на ЭЭГ у большинства таких детей определяется сочетание очаговой, мультирегиональной и диффузной эпилептиформной активности с нарастанием во сне. При низкой частоте припадков данные признаки могут отсутствовать. По результатам МРТ головного мозга удается установить признаки диффузной атрофии коры головного мозга и мозжечка, субкортикальных слоев, иногда – увеличение размеров желудочков. Для подтверждения синдрома Драве используется кариотипирование с определением мутации генов SCN1A или GABRG2.
В педиатрии дифференциальная диагностика синдрома Драве проводится с фебрильными судорогами, митохондриальными и дисметаболическими патологиями, доброкачественной миоклонической эпилепсией младенчества, синдромами Леннокса-Гасто и Дозе, другими формами эпилепсии у детей, которые сопровождаются миоклоническими припадками. Практически идентичную клиническую картину имеет мутация гена PCDH19 – эпилепсия с умственной отсталостью, ограниченная женским полом.
Лечение
Синдром Драве – это форма эпилепсии у детей, которая почти не поддается терапии. Основная цель лечения – снизить чистоту приступов, профилактировать их трансформацию в эпилептический статус. Как правило, большинство распространенных противоэпилептических средств при тяжелой миоклонической эпилепсии младенчества неэффективны. В качестве стартовой терапии показаны вальпроаты (вальпроева кислота) и сульфат-замещенные моносахариды (топирамат). Также могут применяться фармакологические средства из групп барбитуратов и бензодиазепинов. В некоторых случаях при синдроме Драве позитивная динамика отмечается на фоне кетогенной диеты, которая подразумевает большое количество жиров и строгое ограничение углеводов.
Прогноз для жизни при синдроме Драве сомнительный, для выздоровления – неблагоприятный. Дефицит интеллекта, расстройства психики, эпилептические припадки и неврологические нарушения обычно сохраняются на протяжении всей жизни человека, что обусловливает его полную социальную дезадаптацию. Обычно приступы возникают в ночное время или сразу после пробуждения, а их интенсивность и частота уменьшаются. Смертность составляет порядка 15,9-18%. Основные причины – синдром внезапной детской смерти при эпилепсии, интеркуррентные инфекционные заболевания, несчастные случаи во время припадков.
Антенатальная профилактика синдрома Драве аналогична другим наследственным заболеваниям. Она подразумевает медико-генетическое консультирование и планирование беременности, кариотипирование плода посредством амнио- или кордоцентеза. Постнатальные превентивные меры включают в себя исключение гипертермических состояний у ребенка (раннее лечение инфекционных заболеваний, избегание горячих ванн ) и других факторов, которые могут спровоцировать приступ.


